
Nitrosamine Impurities in Pharmaceuticals: Risks, Regulations, and Control
Introduction to Nitrosamine Impurities
Nitrosamine Impurities in Pharmaceuticals: Risks, Regulations, and Control has become one of the most critical quality and patient safety topics in the pharmaceutical industry. Since multiple high-profile drug recalls, regulatory agencies worldwide have intensified their scrutiny of nitrosamines in active pharmaceutical ingredients (APIs) and finished dosage forms.
Nitrosamines are not new contaminants; however, their presence in medicines raised global alarm due to their potent carcinogenic nature even at extremely low exposure levels. As a result, pharmaceutical manufacturers must now proactively identify, evaluate, and control nitrosamine risks throughout the product lifecycle
What Are Nitrosamines?
Nitrosamines are a class of chemical compounds formed when secondary or tertiary amines react with nitrosating agents such as nitrites under certain conditions. These reactions can occur unintentionally during pharmaceutical manufacturing or storage.
Common nitrosamines of concern include NDMA and NDEA, which have been widely studied due to their strong links to cancer in animal studies. Even trace amounts can pose long-term health risks, making strict control essential.
Why Nitrosamines Are a Concern in Pharmaceuticals
The primary concern lies in their genotoxic and carcinogenic properties. Unlike many impurities that have defined safe thresholds, nitrosamines often require control at parts-per-billion (ppb) levels. This creates significant challenges for analytical detection, process design, and quality assurance.
Additionally, patients may take certain medications daily for years, increasing cumulative exposure risk if nitrosamines are present.
Sources of Nitrosamine Impurities in Drug Product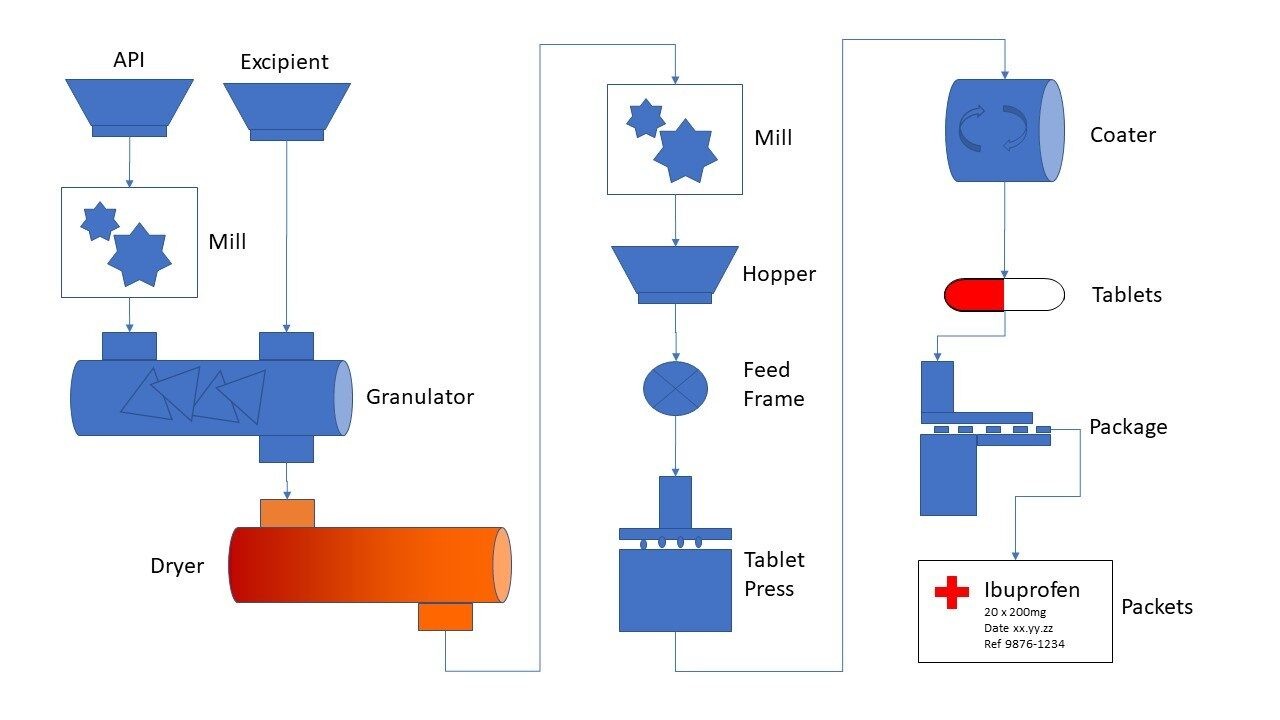
Manufacturing Process-Related Sources
Nitrosamines can form during chemical synthesis when amine-containing intermediates are exposed to nitrosating conditions. Changes in process steps, recycling of solvents, or inadequate cleaning procedures can all contribute.
Raw Materials and Solvent Contributions
Contaminated raw materials, reagents, or recovered solvents may already contain nitrosamines or precursors. Without proper qualification and testing, these materials can introduce impurities into the final product.
Packaging and Storage Factors
Certain packaging materials, inks, or environmental conditions such as high humidity and heat can promote nitrosamine formation during storage, particularly for solid oral dosage forms.
Health Risks Associated with Nitrosamine Exposure
Carcinogenic Potential
Nitrosamines are classified as probable or known human carcinogens based on extensive toxicological data. Long-term exposure has been associated with cancers of the liver, stomach, and esophagus in animal models.
Acceptable Intake Limits
Regulatory authorities establish acceptable intake (AI) limits based on lifetime exposure assumptions. These limits are extremely low, reflecting a one-in-100,000 cancer risk over a lifetime.
Regulatory Landscape for Nitrosamine Control
US FDA Guidelines and Expectations
The U.S. Food and Drug Administration requires manufacturers to:
- Conduct risk assessments for nitrosamines
- Test products where risk is identified
- Implement controls and report findings promptly
- Failure to comply can result in recalls, warning letters, or import alerts.
EMA and European Union Regulations
The European Medicines Agency mandates a stepwise approach involving risk evaluation, confirmatory testing, and risk mitigation. Marketing authorization holders are responsible for ensuring compliance across their supply chains.
Global Harmonization Efforts
International collaboration aims to align acceptable intake limits, testing methodologies, and reporting requirements to reduce regulatory burden while maintaining patient safety.
Detection and Analytical Methods
Chromatographic Techniques
Gas chromatography (GC) and liquid chromatography (LC) are commonly used to separate nitrosamines from complex matrices.
Mass Spectrometry-Based Approaches
Coupling chromatography with mass spectrometry (GC-MS or LC-MS/MS) allows detection at ultra-trace levels, meeting stringent regulatory expectations.
Risk Assessment and Control Strategies
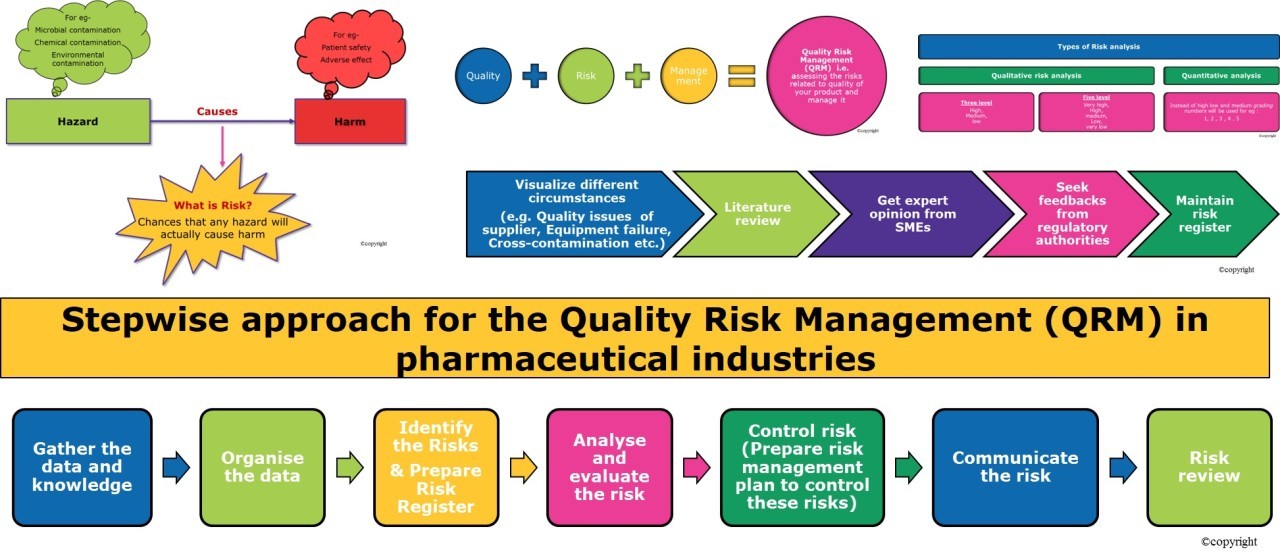
Risk Evaluation During Drug Development
Risk assessment should begin early, evaluating chemical structures, synthesis routes, and degradation pathways that may lead to nitrosamine formation.
Mitigation and Preventive Measures
Effective controls include:
- Reformulating synthesis routes
- Eliminating nitrosating agents
- Improving solvent quality
- Enhancing cleaning validation
Quality Management and Lifecycle Control
Nitrosamine control is not a one-time activity. Continuous monitoring, change management, supplier qualification, and periodic reassessment are essential to ensure long-term compliance and patient safety.
FAQs on Nitrosamine Impurities in Pharmaceuticals
1. Why are nitrosamines regulated so strictly?
Because they are potent carcinogens even at extremely low levels.
2. Can nitrosamines form after product release?
Yes, under certain storage or packaging conditions.
3. Are all nitrosamines equally dangerous?
No, toxicity varies, but most are treated conservatively.
4. Do all drugs need nitrosamine testing?
Only those with identified risk factors.
5. How are acceptable intake limits determined?
They are based on lifetime exposure and cancer risk modeling.
6. Can process changes eliminate nitrosamines completely?
In many cases, yes, with proper design and controls.
Conclusion
Nitrosamine Impurities in Pharmaceuticals: Risks, Regulations, and Control represents a defining challenge for modern pharmaceutical quality systems. Through robust risk assessment, advanced analytical testing, and strict regulatory compliance, manufacturers can protect patients while maintaining trust in the global medicine supply.